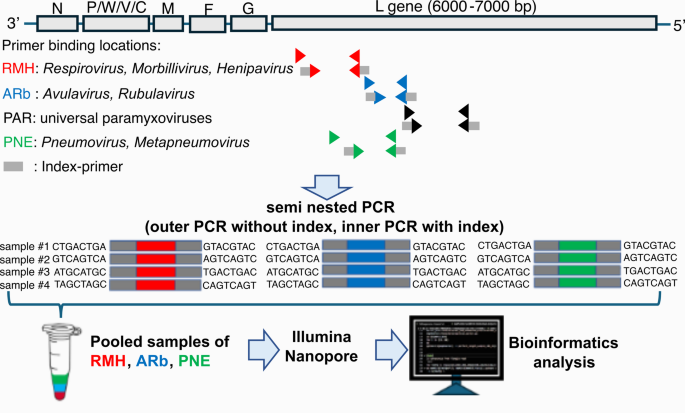
Strong and particular amplification of PCR
The efficacy of the reported PCR amplification system for every paramyxovirus was improved by optimizing the annealing temperature, cycle, and dilution issue of the templates in each outer and interior PCR. Three distinct exams encompass the semi-nested RT-PCR with RMH primers (RMH PCR) to detect Respirovirus, Morbillivirus, and Henipavirus; the semi-nested RT-PCR with ARb primers (ARb PCR) to detect Avulavirus and Rubulavirus; and the semi-nested RT-PCR with PNE primers (PNE PCR) to detect Pneumovirus and Metapneumovirus. The optimum annealing temperature of the outer PCR was decided to be 52 °C. In the meantime, for the interior PCR, 45 °C was optimum throughout all primer teams (Supplementary Fig. S1a, S1b). The optimum template dilution issue from outer to interior PCR was 100 to 150-fold, and 100-fold was chosen (Supplementary Fig. S2a). The optimum cycles for the outer PCR had been 42 cycles (Supplementary Fig. S2b). These for the interior PCR had been determined to be 32, 32, and 15 cycles for the RMH, ARb, and PNE PCR, respectively, contemplating elevated non-specific amplification of PNE noticed with 32 cycles (Supplementary Fig. S2b).
Dedication of sensitivity and specificity of semi-nested PCR
The sensitivity of the semi-nested PCR system was validated via serial dilution of the plasmid templates and twofold replication of the impartial experiment (Supplementary Fig. S3). The gel electrophoresis picture of the amplicons indicated that the restrict of detection for RMH PCR focusing on the plasmid coding goal area from Human parainfluenza 3, Canine distemper, Cedar virus, and Salem virus is 1–10 copies/response. Alternatively, ARb PCR was in a position to detect a minimal of 100-1,000 copies/response for Avian paramyxovirus 3 and Simian virus 5, whereas PNE PCR confirmed a detection restrict of 10–100 copies/response for Human metapneumovirus and Bovine respiratory syncytial virus. The specificity of RMH, ARb, and PNE PCR was validated utilizing a plasmid with viral sequence insertion from 8 representatives’ viruses belonging to all genera of paramyxoviruses. As proven in Supplementary Fig. S4, every PCR system particularly amplified the plasmid from its goal group. The RMH PCR amplified plasmids from Human parainfluenza 3, Canine distemper, Cedar virus, and Salem virus, ARb PCR amplified plasmid from Avian paramyxovirus 3 and Simian virus 5, and PNE PCR amplified plasmids from Human metapneumovirus and Bovine respiratory syncytial virus.
Detection of the sequences of paramyxoviruses in bats utilizing semi-nested RT-PCR and NGS evaluation
A complete of 135 wild bats belonging to 4 households and 22 species (Pteropodidae (n = 103), Rhinolophidae (n = 20), Vespertilionidae (n = 3), and Hipposideridae (n = 9)) had been collected from 4 islands in Indonesia: Java island (Banyumas (n = 25), Gunungkidul (n = 22), Banyuwangi (n = 4)), Sulawesi island (Tomohon (n = 31)), Flores island (Labuan Bajo (n = 27)); and Nusa Penida island (n = 26) (Desk 1). Amplification was noticed clearly from 8/270 samples by gel electrophoresis from ARb PCR (Fig. 2; Desk 2, Supplementary Fig. S5b). In distinction, the precise amplicon was not noticed with RMH and PNE PCR (Supplementary Fig. S5a and c). However, all amplicons had been subjected to next-generation sequencing, together with constructive and damaging samples from RMH, ARb, and PNE PCR. The outcomes revealed the presence of viral sequences belonging to the Paramyxoviridae household within the ARb amplicons, thus, solely this amplicon was additional characterised with deep sequencing.

Gel electrophoresis photos of ARb PCR amplicons.
The anticipated amplicon dimension (244 bp) from the eight seen bands was decided. Seven samples had been confirmed by NGS (proven in pink arrowheads). One pattern was synthetic amplification (proven in inexperienced arrowhead). 5 samples (85, 91, 170, 180, and 197) exhibited a powerful band, whereas three (218, 237, and 263) displayed a weaker band. The combination of PCR with NGS enhanced the sensitivity of the system. Viral reads had been present in one other 22 ARb amplicons with invisible bands utilizing the Illumina (proven in blue arrowheads). The unique photos are proven in Supplementary Fig. S5b.
In depth NGS evaluation utilizing Illumina Miseq platforms efficiently detected viral sequences in seven of eight ARb amplicons with seen bands by gel electrophoresis on the anticipated dimension (Fig. 2). One pattern (T-NP01) with a weak band was damaging, indicating non-specific amplification. Alternatively, viral reads had been present in one other 22 ARb amplicons with invisible bands in agarose gel electrophoresis. Equally, by flongle-Nanopore, viral sequences had been present in seven of eight ARb amplicons with a visual band and one other 4 ARb amplicons with an invisible band (Fig. 2; Desk 2). Amongst them, 17 to 66,544 and seven to 160 reads for the Illumina and the Nanopore dataset, respectively, had been assigned as viral origin based mostly on blastn homology evaluation (Desk 2). Right here, they had been solely thought of constructive when amplicons had been confirmed within the gel, and viral reads had been obtained to keep away from the potential for false positives. Extra particularly, seven samples (T-NP19, R-NP19, R-GK013, R-GK019, T-LB06, R-LB06, and T-LB16) had been considered constructive. The constructive samples had a better learn quantity, endorsing the reliability of the standards. Alternatively, samples through which viral reads had been detected however amplicons couldn’t be confirmed within the gel had been conservatively thought of damaging to keep away from resulting in a false conclusion concerning the bat’s virome. As a result of attainable artifacts delivered from cross-contamination, index-hopping from sequencing errors, and template switching throughout library preparation couldn’t be excluded.
The recognized viral sequences exhibited a comparatively low identification, starting from 80.85 to 82.44% when in comparison with these within the NCBI nt database. Based mostly on blastn hit outcomes, these viral sequences had been near Achimota virus 1, Achimota virus 2, and Paramyxovirus bat/GH18/2008 (Desk 2). The Achimota virus 1-like sequences had been detected in each tracheal and rectal swabs from Eonycteris spelaea. The Achimota-virus 2-like sequences had been detected in three rectal swabs and one tracheal swab from Cynopterus brachyotis. The Paramyxovirus bat/GH18/2008-like sequences had been recognized in a single tracheal swab from Rousettus amplexicaudatus. Constantly, 100% an identical sequences had been present in each rectal and tracheal swab samples from the identical bat: R-NP19/T-NP19 and R-LB06/TLB06.
Validation by one other pan-paramyxovirus PCR system
To endorse the 31 viral sequences from 29 samples recognized by the ARb PCR (Desk 2), one other identified pan-paramyxovirus semi-nested RT-PCR, so-called PAR PCR, was utilized. The PAR PCR targets different areas of the L gene from the paramyxovirus’s viral genome (Fig. 1). The seen amplicon was proven in 4 samples by gel electrophoresis (Supplementary Fig. S6). The choice was made to sequence all amplicons utilizing the nanopore platform, whether or not the bands had been seen or not. Lastly, paramyxovirus-like sequences had been recognized solely from the 4 PCR-positive samples, and the outcomes of the blastn hit with nt NCBI database are described under. The tracheal and rectal samples from the identical bat T-LB06 and R-LB06 had been recognized as much like Human orthorubulavirus 2, genus Orthorubulavirus, with 70.88% similarity (Desk 2). As well as, the sequences obtained by ARb PCR from the identical bat LB06 had been recognized as much like one other Orthorubulavirus member, Achimota virus 1 (Desk 2). From the tracheal pattern, T-LB16, the viral sequence was recognized as much like Tuhoko virus 3, genus Pararubulavirus, with 75.87% similarity (Desk 2). The sequences obtained by ARb PCR from the identical bat pattern had been much like Paramyxovirus bat/GH18/2008, unclassified Paramyxoviridae, with 80.85% similarity. Apparently, the sequences derived from a tracheal pattern, T-NP19, had been discovered to be inconsistent with the ARb-PCR outcomes. The sequence from the PAR amplicon was recognized as much like Henipavirus hendraense, genus Henipavirus, with 73.12% similarity (Desk 2). Nevertheless, the sequences obtained by ARb PCR had been much like Achimota virus 2, genus Pararubulavirus.
Validation by Bridge PCR
The sequences from two separate viral genome places had been recognized by ARb and PAR PCR. To additional characterize if these two recognized sequences had been from the identical viral genome or not, RT-PCR throughout each ARb and PAR areas was performed (Supplementary Fig. S7f). In 4 samples, T-LB06, R-LB06, T-LB16, and T-NP19, which had been each constructive for ARb and PAR-PCR, had been characterised. It was initially performed utilizing AVU-RUB-F1 and PAR-R primers as outer PCR adopted by ARb-F2-index and PAR-R primers as interior PCR. The anticipated amplicon was efficiently obtained for one pattern, T-LB16 (Supplementary Fig. S7a). Amplicon was sequenced by the Sanger sequence, and within the ARb area and PAR area, the identical sequence was noticed in comparison with the sequence obtained by ARb PCR and PAR PCR. A blast search utilizing the entire sequence area throughout ARb-F2-index and PAR-R primer area recognized Tuhoko virus 3, genus Pararubulavirus, as probably the most related sequence with 75.99% identification (Desk 2). For the remaining three samples, one other nested PCR utilizing particular primers designed based mostly on the obtained ARb and PAR sequences was tried. The anticipated amplicons had been efficiently obtained from two samples (T-LB06 and R-LB06) utilizing ARb-F1.C4, PAR-R1.C6, ARb-F2.C4, and PAR-R2.C6 primers (Supplementary Desk S1). Each samples confirmed the anticipated 570 bp band dimension (Supplementary Fig. S7b), and their sequences had been most much like Human orthorubulavirus 2, genus Orthorubulavirus, with 71.26% identification (Desk 2). The obtained sequences overlapping the area of ARb and PAR-PCR had been the identical as these obtained by NGS sequences. In distinction, no amplicon was obtained from T-NP19 with any primer mixture (Supplementary Fig. S7c, d, and e).
Phylogeny and phylogeography amongst paramyxovirus-like and bats
A phylogenetic evaluation of the viral sequences obtained by ARb PCR recognized 4 distinct sequences belonging to Pararubulavirus and Orthorubulavirus (Fig. 3a). From the ARb amplicons, a brand new clade of PQ784049.1 Pararubulavirus/T-NP19 and PQ784049.1* Pararubulavirus/R-NP19 was generated. The asterisks (*) point out that the sequences obtained are an identical to these registered within the GenBank. One other clade was generated from PQ784050.1 Pararubulavirus/R-GK013 and PQ784050.1* Pararubulavirus/R-GK019. These sequences had been intently associated to Achimota virus 2, and Cynopterus brachyotis was recognized as a bunch. Moreover, one other clade near Tuhoko virus 3 was generated from PQ784051.1 Orthorubulavirus/T-LB16, and this sequence was detected in Rousettus amplexicaudatus. Furthermore, one other clade from PQ784052.1 Orthorubulavirus/R-LB06 and PQ784052.1* Orthorubulavirus/T-LB06, don’t share with particular identified reference viruses. These sequences had been intently associated to Mumps orthorubulavirus, and Eonycteris spelaea had been detected as hosts.

Phylogeny amongst paramyxoviruses-like sequences from bats and associated viral sequences. The utmost-likelihood phylogenetic tree was constructed utilizing (a) ARb area and (b) PAR area. The uncooked learn variety of sequences from every pattern is proven inside parentheses. Bootstrap values from SH-aLRt calculated by 1000 replicates are proven as black circles over 50%. Purple strains present the ARb and PAR sequence pair, which had been validated to be derived from the identical viral genome by bridge-PCR. Host bat species are proven as coloured bat figures subsequent to the sequence ID within the phylogeny. The asterisks (*) had been added if an identical sequences registered in GenBank had been obtained.
The viral sequences from PAR amplicons had been recognized from 4 examined samples and categorised as Henipavirus, Pararubulavirus, and Orthorubulavirus (Fig. 3b). A viral sequence of PQ784046.1 Henipavirus/T-NP19 was recognized near Nipah virus and fashioned a brand new department along with members of Henipavirus genus. This sequence originated from Cynopterus brachyotis. In distinction, with the identical pattern, viral sequence from ARb amplicon (PQ784049.1 Pararubulavirus/T-NP19) was detected as intently associated to a special genus, Pararubulavirus. This means that the sequences had been derived from completely different viral genomes, a conclusion additional supported by the proof that the bridge amplicon from ARb and PAR was not efficiently obtained utilizing typical RT-PCR (Supplementary Fig. S7c, d, and e). Moreover, a sequence of PQ784048.1 Orthorubulavirus/T-LB16 was derived from the PAR amplicon, and Rousettus amplexicaudatus was obtained as a bunch. The sequence was decided in the identical clade as Tuhoko virus 3 and categorised as a member of Pararubulavirus much like a viral sequence derived from the ARb amplicon. This means that the viral sequences had been obtained from the identical viral genome. The bridge amplicon from them lends additional assist to this speculation (Desk 2; Fig. 3a and b). Moreover, a brand new clade was generated from PQ784047.1 Orthorubulavirus/R-LB06 and PQ784047.1* Orthorubulavirus/T-LB06. Simian virus 41 was probably the most intently associated to this sequence, along with one other virus member from the Orthorubulavirus. With an identical pattern, a viral sequence from the ARb amplicon (PQ784052.1 Orthorubulavirus/R-LB06 and PQ784052.1* Orthorubulavirus/T-LB06) was categorised as Orthorubulavirus, according to the classification of the bridge amplicon sequence from the identical samples (Desk 2; Fig. 3a and b). This proof signifies that these viruses had been recognized from the identical viral genome.
The geographical distribution of paramyxovirus-like sequences was examined throughout six places (Fig. 4). The sequences had been recognized and confirmed in three of the six places, with Banyumas being the distinctive location the place no paramyxovirus-like sequence was detected. Different places, Banyuwangi and Tomohon, had been decided as areas with false constructive outcomes. The detection charges had been calculated by evaluating the variety of bats with recognized viral sequences to the entire variety of collected bats from every location. The detection charge assorted among the many areas, with the very best charge noticed in Gunungkidul 2/22 bats (0.09%), adopted by Labuan Bajo 2/27 bats (0.07%), and Nusa Penida 1/26 bats (0.03%). Furthermore, a variation of bat species recognized from these viral sequences was noticed in Labuan Bajo. Notably, Pararubulavirus-like demonstrated a wider distribution in comparison with different teams, which had been detected from Gunungkidul and Nusa Penida. Viral diversities had been demonstrated in bats from Nusa Penida and Labuan Bajo.

The distribution of paramyxoviruses-like in collected bats from Indonesia. The variety of bats with detected viral sequences was in contrast with collected bats from every location, as proven on this determine. The distribution of viral sequences amongst hosts was described in coloring bats, with the places the place the hosts had been captured. The viral sequences from ARb and PAR amplicons, categorised based on phylogenetic relationship in Fig. 3, had been proven with completely different colours. The places of detected viral sequences are indicated with pink dots, and damaging outcomes with black dots. This map was supplied by GADM and subsequently organized by QGIS 3.40 Bratislava.