Biomaterials
Human organic samples have been obtained from Discovery Life Sciences. The analysis use of those specimens was accredited by the corresponding institutional evaluate boards beneath protocol numbers DLS-BB044-v.1 and SB-GP_v1. All sufferers supplied written knowledgeable consent to Discovery Life Sciences for the analysis use of their organic samples.
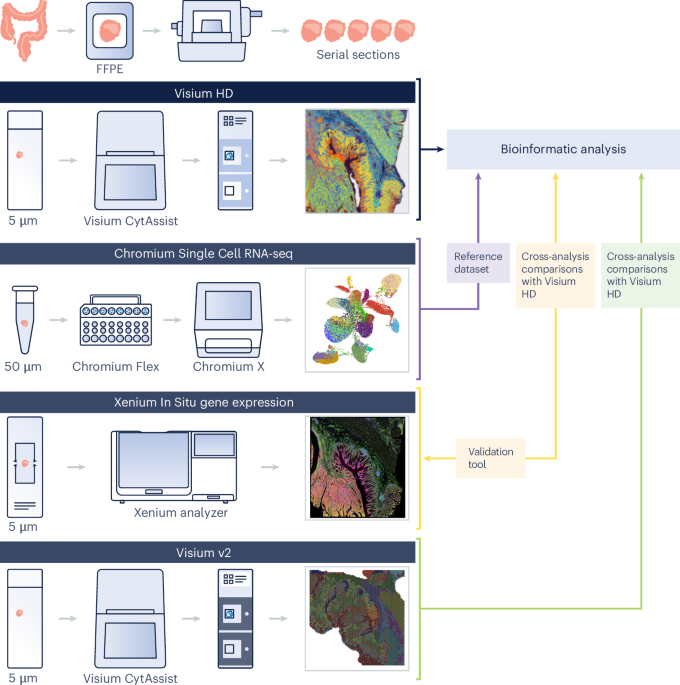
An summary of the experimental design is introduced in Fig. 1. Samples from 5 sufferers with colon adenocarcinoma (two males, three females, ages 58–72, pretreatment) have been included on this examine. From every affected person, we included CRC FFPE blocks and NAT blocks for a subset of three sufferers (bringing the full to eight FFPE blocks), and we obtained paired freshly frozen dissociated tumor cells (for immune profiling evaluation), alongside the three chosen FFPE blocks used for spatial profiling (Desk 1).
Tissue sectioning
Adjoining or near-adjacent 5-µm sections have been taken from the FFPE tissue blocks following the Xenium In Situ for FFPE — Tissue Preparation Information (CG000578, revision C) for the Xenium workflow or the Visium CytAssist Spatial Gene Expression for FFPE — Tissue Preparation Information (CG000518, revision C) for the Visium workflows.
Visium HD Spatial Gene Expression
H&E staining and imaging have been carried out following the Visium HD FFPE Tissue Preparation Handbook (CG000684). Samples have been then processed and sequenced following the Visium HD Spatial Gene Expression Reagent Kits Person Information (CG000685). Area Ranger model 3.0 was used for evaluation. Along with the native 2-µm function dimension, Area Ranger outputs Visium HD knowledge binned to 8-µm and 16-µm decision. Except in any other case specified, downstream analyses have been carried out on the 8-µm decision knowledge, which give a 16-fold improve within the common variety of UMI reads per bin within the gene–barcode matrix in comparison with a 2-µm sq. (Visium HD Spatial Gene Expression Efficiency Technical Word, CG000686, revision A), which is of an affordable dimension to be processed by means of third-party instruments. Moreover, the 8-µm bin knowledge present sturdy cell kind annotations (that’s, bins are sufficiently small to supply single-cell scale for many cell varieties). We merged the UMI depend matrices of the three sections and, given the dataset’s giant dimension, we adopted the sketch-based evaluation method in Seurat (https://satijalab.org/seurat/articles/seurat5_sketch_analysis), sampling 15% of your complete dataset for downstream evaluation52,53. The 15% sampling fee was chosen to make sure environment friendly computational efficiency. We used the leverage rating because the sketching technique. This technique oversamples uncommon populations to protect the organic complexity of the pattern. We recognized variable options, scaled the info, carried out principal-component (PC) evaluation (PCA) and carried out graph-based clustering (20 PCs, decision = 0.8). DGE evaluation recognized cluster marker genes, enabling guide annotation into ten broad cell varieties (stage 1). For stage 2 annotations, we reclustered every stage 1 cluster (25 PCs, decision = 0.1) to refine subtypes, carried out DGE and annotated cell varieties. Last annotations have been prolonged to the total dataset.
When wanted, the two × 2-µm knowledge have been used (together with nuclear stain) to supply extra info, significantly to find smaller cell varieties (Nuclear segmentation).
Spatial accuracy
To measure spatial accuracy of mRNA detection, we recognized morphologically distinct ROIs after which pinpointed marker genes distinctive to every ROI. These marker genes ought to be expressed solely in squares instantly beneath their corresponding ROI. Utilizing QuPath model 0.4.4 (ref. 54), we chosen 4 ROIs inside regular colon mucosal glands (hereafter known as ‘supply masks’) and areas of adjoining muscularis mucosae (‘adjoining masks’), selecting three goblet cell marker genes (CLCA1, FCGBP and MUC2). We mapped the places of all transcripts for these marker genes and calculated the proportion of precisely localized transcripts for every of the 4 ROIs. For the remaining transcripts, we decided the Euclidean distance from the sting of the closest supply masks to the sq. of transcript detection. Moreover, we calculated the densities of the marker genes inside each the supply and adjoining masks for every ROI.
Visium CytAssist Spatial Gene Expression for FFPE
Visium CytAssist Spatial Gene Expression for FFPE (‘Visium v2’) was run on a subset of samples to exhibit technological enhancements of Visium HD. FFPE serial sections (5 µm) have been positioned on commonplace glass slides and stained with H&E following the Demonstrated Protocol Visium CytAssist Spatial Gene Expression for FFPE — Deparaffinization, H&E Staining, Imaging & Decrosslinking (CG000520). Samples have been processed and sequenced following the Visium CytAssist Spatial Gene Expression Reagent Kits Person Information (CG000495). Area Ranger model 3.0 was used for evaluation.
Sensitivity comparability of Visium v2 and Visium HD
We assessed the sensitivity of Visium HD in comparison with Visium v2 on a gene-by-gene foundation. Matched areas have been manually chosen in Loupe Browser model 8.0, and probe–barcode matrices from every dataset, generated by Area Ranger, have been imported into Seurat model 5 (ref. 52) utilizing the read10xh5() perform. We used the ggplot2 R package deal to graph per-probe UMI counts. The info have been displayed on a log10 + 1 scale, with Spearman correlations calculated as r2. Our comparability spanned all probes throughout your complete transcriptome and particularly targeted on probes crossing an exon–exon splice junction (7,605 of 54,580 probes). This latter comparability helps exclude most probes that might goal gDNA or be inclined to off-target results. These probes which can be marked as doubtlessly binding to gDNA have been excluded from the evaluation computationally.
The gDNA estimate is obtained by becoming a piecewise linear mannequin to genes with each spliced and unspliced probes. The mannequin predicts the log-transformed gene counts for unspliced probes as a perform of log-transformed gene counts for spliced probes and log-transformed estimated gDNA counts per gene per 1,000 spots beneath tissue:
$${{rm{x}}}_{{rm{unspliced}}}={rm{gDNA}}+Bleft({{rm{x}}}_{{rm{spliced}}}proper)-{hat{{rm{X}}}}_{{rm{gDNA}}}{rm{II}}left({{rm{x}}}_{{rm{spliced}}} > {hat{{rm{X}}}}_{{rm{gDNA}}}proper),$$
the place X̂gDNA is the estimated gDNA UMIs per gene, xunspliced and xspliced are the common variety of UMIs equivalent to unspliced and spliced probes from a single gene, respectively, and II(({{rm{x}}}_{{rm{spliced}}} > {hat{{rm{X}}}}_{{rm{gDNA}}})) is an indicator perform that equals one if xspliced is larger than ({hat{{rm{X}}}}_{{rm{gDNA}}}) and 0 in any other case.
For any fastened estimate of ({hat{{rm{X}}}}_{{rm{gDNA}}}), the mannequin parameters gDNA and B are estimated by becoming a linear regression with a two-dimensional parameter vector. The related ({hat{{rm{X}}}}_{{rm{gDNA}}}) values to contemplate whereas becoming the mannequin are solely these xspliced values seen within the genes into account. Subsequently, a linear mannequin with each xspliced worth seen within the dataset as ({hat{{rm{X}}}}_{{rm{gDNA}}}) is match, and the mannequin with the smallest residual sum of squares is used for the estimates. For extra particulars on gDNA, see the technical word Visium CytAssist Spatial Gene Expression for FFPE: Strong Knowledge Evaluation with Minimal Impression of Genomic DNA (CG000605, revision A).
Chromium Single Cell Gene Expression Flex
Cells have been dissociated from 50-µm FFPE curls utilizing the Demonstrated Protocol for Isolation of Cells from FFPE Tissue Sections for Chromium Fastened RNA Profiling (CG000632). Library preparation and sequencing adopted the Chromium Fastened RNA Profiling for Multiplexed Samples Person Information (CG000527, revision D). Cell Ranger model 8.0.0 was used for evaluation. To construct the atlas, we used Seurat model 5 (ref. 52) to import the H5 file produced by the cellranger aggr pipeline. We then plotted the distribution of UMIs and genes per barcode, excluding the highest and backside 2.5% of the distribution to account for outliers. We then used the identical sketch method because the HD knowledge: recognized variable options, scaled the info, carried out PCA and carried out graph-based clustering (25 PCs, decision = 0.6) and carried out DGE evaluation to determine cluster marker genes, enabling guide annotation into ten broad cell varieties (stage 1). For stage 2 annotations, we reclustered every stage 1 cluster (25 PCs, decision = 0.1) to refine subtypes, carried out DGE and annotated cell varieties. Last annotations have been prolonged to the total dataset.
Spot deconvolution
Deconvolution strategies purpose to determine the cell varieties and their relative proportions contributing to the sign captured in a particular space (spot, sq. or bin). Spot deconvolution was used to categorise and label bins with cell varieties derived from the Single Cell Atlas. We ran spacexr55 utilizing doublet mode, which assigns one to 2 cell varieties per spot and is really helpful for applied sciences with excessive spatial decision akin to Visium HD. Among the many outputs, this mode supplies the highest two ranked cell varieties per spot, a matrix with the weights for all cell varieties within the reference in every of the spots and a variable representing the category of each bin. The accessible courses are ‘singlet’ (one cell kind), ‘doublet_certain’ (two cell varieties), ‘doublet_uncertain’ (two cell varieties, however solely assured of 1), ‘reject’ (no prediction given). See the spacexr vignette for extra element (https://uncooked.githack.com/dmcable/spacexr/grasp/vignettes/spatial-transcriptomics.html). Owing to the elevated variety of barcoded squares in Visium HD, we modified the code to enhance runtime (https://github.com/dmcable/spacexr/pull/206). We chosen a minimal UMI threshold of 100 for a bin to be thought of for the deconvolution step.
Characterizing the tumor periphery
To determine and analyze tumor periphery areas, we developed a customized pipeline (https://github.com/10XGenomics/HumanColonCancer_VisiumHD). The important thing benefit of this technique is its versatility; it isn’t restricted to tumors and may be utilized to determine cell varieties within the boundaries of different tissues, akin to colon glands or skeletal muscle tissue. Our algorithm first selects all bins labeled with the given cluster or cell kind. For every bin, the algorithm then identifies all neighboring bins inside a user-defined radius. We outlined this as 50 µm primarily based on organic relevance and to take advantage of the excessive decision of Visium HD. A 50-µm boundary evaluation isn’t potential with Visium v2, which has a spot diameter of 55 µm. To precisely delineate the boundary of the cluster or cell kind, the algorithm excludes bins with the identical label because the preliminary bin. To additional refine the boundary identification, we eliminated singlet bins labeled with the given cluster however not in proximity to different bins with the identical label. Particularly, we excluded bins that had fewer than 25 neighboring bins with the identical label to make sure the number of well-defined areas throughout the tissue.
For the macrophage-specific evaluation, we chosen 8-µm bins that have been deconvolved as macrophages and situated inside 50 µm of tumor areas. For every affected person, variable options have been recognized, the info have been scaled and PCA was carried out. Graph-based clustering was then carried out utilizing the highest ten PCs with a decision of 0.2. Clusters have been annotated primarily based on the expression of SPP1 and SELENOP, and an built-in dataset was generated, together with solely the macrophage subpopulations persistently recognized throughout all sections. To increase this evaluation, all bins labeled as macrophages throughout all sections have been included, regardless of their proximity to the tumor boundary. For this broader dataset, 15 PCs and a decision of 0.2 have been used for clustering to realize better resolving energy throughout sufferers. This method aimed to find out whether or not the recognized subpopulations have been shared throughout sufferers or particular to particular person sufferers.
Distance, native and regional evaluation
After figuring out areas enriched with immune cells, we chosen the highest three areas exhibiting the best macrophage density (inside bins categorized as tumor tissue) and delineated these areas as ROIs with a radius of 350 µm. Moreover, we recognized a ‘chilly’ area missing immune infiltration. To determine genes related to elevated immune cell density, we used DGE evaluation. Subsequent enrichment evaluation of differentially expressed genes (ranked by log2 (fold change)) used the Hallmark gene units linked to particular organic pathways40 (https://www.gsea-msigdb.org/gsea/msigdb/human/genesets.jsp?assortment=H).
Ligand–receptor evaluation
To evaluate cell–cell communication between macrophages and T cells, we used LIANA model 0.1.12, which supplies a framework to mix cell communication strategies to deduce ligand–receptor interactions33. We chosen all bins throughout the beforehand outlined tumor periphery area (inside 50 µm of the tumor boundary) to create an impartial Seurat object for use as enter. We filtered the expected interactions by deciding on any interplay with aggregate_rank ≤ 0.01. This process was finished on a per-patient foundation.
Immune density evaluation
To map the places of particular immune cell varieties within the CRC samples, we used the coordinates for every bin and their labels supplied by deconvolution. We counted solely bins that have been labeled as singlets. We used 2D kernel density estimation to pick areas enriched in a given cell kind. Density values have been scaled to a most of 1.
Nuclear segmentation
Utilizing the 8 × 8-μm binned knowledge for the periphery evaluation meant that the majority T cells within the tumor periphery have been assigned to doublet bins or rejected (the algorithm was unable to foretell the cell kind) and subsequently excluded, making them difficult to spatially localize (Fig. 6a and Prolonged Knowledge Fig. 8). That is an impact of the 8 × 8-μm binned knowledge method for cell project (Fig. 6b), wherein smaller cell varieties akin to T cells (as in comparison with tumor cells) might colocalize different cell varieties throughout the 8 × 8-μm bin. To beat this, we used an alternate method to assign 2 × 2-μm squares to cells. We first recognized areas enriched in both CD4+ or CD8+ T cells, impartial of whether or not they have been assigned to a singlet or doublet bin, and, with using the nuclear stain from a high-resolution H&E microscope picture, we carried out nuclear segmentation on these areas. To section nuclei from the H&E pictures and assign 2 × 2-µm bins to the recognized nuclei, we adopted the evaluation information ‘Nuclei Segmentation and Customized Binning of Visium HD Gene Expression Knowledge’ (https://www.10xgenomics.com/analysis-guides/segmentation-visium-hd). The segmentation process was run on the total part utilizing StarDist56. We used affine transformations to protect the segmentation polygons when subsetting the picture to particular ROIs. As soon as the 2-µm bins have been assigned to the corresponding nuclear polygons, the info have been remodeled to create a gene-by-nucleus UMI depend matrix for additional processing.
Single-cell immune profiling
Freshly frozen dissociated tumor cells have been sorted primarily based on CD45 and CD3 expression to isolate T cells. Gene expression (5′) and TCR libraries have been generated and sequenced following the Chromium Subsequent GEM Single Cell 5′ Reagent Kits v2 (Twin Index) Person Information (CG000331). Cell Ranger model 8.0.0 was used for evaluation.
Design of TCR clonotype probes
Customized probes for Xenium have been developed to focus on three CDR3 sequences recognized by VDJ sequencing (full record of clonotypes in Supplementary Desk 4, probe sequences in Supplementary Desk 3). One 40-bp probe was designed for every CDR3, centered on the CDR3 with some overhang into the adjoining framework areas. These probes have been designed following the specs within the Species Standalone Customized and Superior Customized Panel Design for Xenium In Situ Technical Word (CG000683, revision C).
Xenium In Situ
The P1CRC, P2CRC and P5CRC samples have been processed following the Xenium In Situ Gene Expression with Morphology-based Cell Segmentation Staining Person Information (CG000749) with the Xenium Human Colon Gene Expression Panel (322 genes) supplemented with a further 100 genes chosen to determine numerous immune populations we noticed within the Visium HD knowledge and characterize the TME (for the whole gene record, see Supplementary Desk 2). The panel was designed utilizing Xenium Panel Designer following the steering within the Xenium Add-on Panel Design Technical Word (CG000643, revision B). The Xenium Onboard Evaluation pipeline model 2.0.0 was run instantly on the instrument for picture processing, cell segmentation, picture registration, decoding, deduplication and secondary evaluation.
Reporting abstract
Additional info on analysis design is out there within the Nature Portfolio Reporting Abstract linked to this text.