Linear ion traps (LITs) are strong, delicate, and quick mass analyzers, but these devices have restricted mass decision. Beforehand, our lab demonstrated that LITs may very well be used successfully as stand-alone mass analyzers to measure low-input samples utilizing an Orbitrap Eclipse™ Tribrid mass spectrometer22. In that work, we detected roughly 400 proteins from single cells utilizing data-independent acquisition coupled with chromatogram libraries to assist make detections23. Whereas our Eclipse instrument configuration ignored the high-resolution Orbitrap mass analyzer, we carried out these experiments within the context of a high-end Tribrid instrument. Moreover, the 400 proteins we measured had been the simplest to look at however not essentially probably the most biologically helpful to observe. Whereas diminished illustration approaches24,25 that quantify a restricted panel of simply noticed proteins may help infer organic states, important hurdles have to be overcome to foretell the expression patterns of unmeasured proteins. As such, immediately measuring panels of proteins of curiosity in low-input samples utilizing focused proteomics could also be preferable to world proteomics.
On this work, we sought to reply three remaining questions. First, by eliminating the Orbitrap, may an inexpensive Q-LIT mass spectrometer carry out at a excessive stage of analytical rigor as a stand-alone instrument for each library era and focused proteomics measurement? Second, can a Q-LIT mass spectrometer quantify peptides at and under the extent of single cells? Third, can quantitative experiments measure low-level, biologically related proteins, resembling cytokines and transcription elements, at or under 1 ng? To this finish, we assessed a number of parameters of a hybrid Q-LIT design produced by Thermo Scientific. First, we examined proteome-wide library era; then, we assessed quantitative linearity utilizing focused PRM experiments with 100, 10, and 1 ng pattern inputs. Lastly, we examined sensitivity and measurement consistency in a organic context.
The Q-LIT platform is a flexible instrument that may carry out world (DIA) and focused (PRM) proteomics with the identical instrument. Devoted low-resolution triple quadrupole devices are able to extremely delicate measurements with broad dynamic ranges. Nonetheless, they’re restricted to chose response monitoring (SRM) for particular precursor/fragment ion transitions. The Sciex QTRAP platform is a hybrid triple quadrupole that may scan as an ion lure within the final quadrupole, making it additionally able to PRM and DDA. Whereas related in geometry to the Stellar, the QTRAP will not be configurable to carry out DIA, partially due to its slower pace. In distinction, Stellar can scan roughly 10× quicker than the QTRAP 6500 + , making it an fascinating candidate for a low-resolution instrument for creating a stand-alone workflow for transitioning world outcomes to focused assays. Different high-resolution devices may also carry out each world and focused scans; due to this fact, we wished to check the Q-LIT to current instrumentation utilizing the Q-Orbitrap (Exploris 480) as a benchmark. Efficiency traits of different associated devices from all kinds of distributors are tabulated in Supplementary Desk 1, which was modified from a latest literature evaluation by Peters-Clarke et al.26. We’ve offered further info on working the Q-LIT in Supplementary Notice 1.
Assessing the efficiency of a Q-LIT mass spectrometer
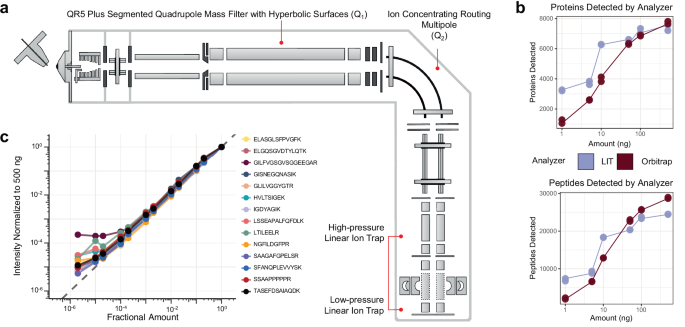
The hybrid Q-LIT mass spectrometer has improved ion transmission options able to performing fast scans as much as 200 kDa/second (Fig. 1a). The instrument shares most of the identical design elements as current Orbitrap-based devices27,28,29. Ions are handed by a mass filter quadrupole (Q1), then concentrated inside the ion routing multipole (Q2) earlier than mass evaluation within the LIT. The Q1 mass filter upstream of the LIT is designed to extend ion transmission utilizing an optimized rod form with hyperbolic surfaces that enable for isolation home windows as small as 0.2 to 2 m/z FWHM (sometimes under 1 m/z)30. Inside this work, we usually keep a minimal of two m/z isolation home windows for PRM to seize a number of isotopes per precursor concurrently, thus rising sensitivity. These configurations produce excessive scanning speeds by performing ion accumulation in parallel with mass evaluation within the low-pressure LIT31,32.

a The instrument schematic of the Q-LIT MS. Ions enter the primary QR5 Plus Segmented Quadrupole Mass Filter with Hyperbolic floor (Q1) earlier than coming into into the Ion Concentrating Routing Multipole (IRM). The IRM behaves because the collision and storage cell. Ions are then moved to the high-pressure cell of the dual-pressure LIT and ultimately to the low-pressure cell for mass evaluation. b The variety of HeLa proteins and peptides detected from 1 to 500 ng inputs analyzed with an Orbitrap (Exploris 480) and LIT (Stellar). Every enter stage was collected in duplicate. c The intensities of PRTC peptides in a HeLa background diluted in water over >5 orders of magnitude. Intensities had been normalized to 500 ng and scaled to 1. The grey dashed line represents a 1 to 1 becoming between the quantity analyzed and the depth acquired.
We first in contrast the Q-LIT to a Q-Orbitrap utilizing wide-window DIA. We analyzed HeLa peptides with mounted 8 m/z isolation home windows (LIT) or 16 m/z staggered isolation home windows33 (Orbitrap, successfully 8 m/z after demultiplexing) at a number of enter ranges. Much like earlier work17,22, we noticed the LIT outperformed the Orbitrap at low enter (Fig. 1b), and we recognized over 6000 proteins (filtered to a 1% FDR) from solely 10 ng of enter materials. With the Q-LIT, the purpose of diminishing returns with DIA peptide and protein detection is 5× greater than we beforehand noticed with the Eclipse21, underlining the advantages of quicker MS2 scanning. At 50 ng or greater enter, the Orbitrap basically equaled or outperformed the LIT, the place entry to excessive mass accuracy grew to become extra essential than elevated sensitivity.
We then wished to evaluate the dynamic vary of focused proteomics utilizing the LIT. To take action, we collected a PRM assay focusing on Pierce Retention Time Calibration (PRTC) peptides spiked right into a HeLa proteome at a ratio of 100 fmol to 100 ng and diluted over 5 orders of magnitude. We acquired the PRTC measurements at 67 kDa/second with a most ion injection time of 200 ms and quantified them in Skyline after normalizing to a 500 ng injection (Fig. 1c). We discovered that the instrument gave a linear sign over 3 orders of magnitude for each PRTC peptide.
A Q-LIT workflow for producing PRM assays utilizing translation libraries
PRM assays are generally generated from varied sources, together with public repositories that retailer focused proteomics knowledge such because the PeptideAtlas34, CPTAC35, or Panorama36. Moreover, assays might be constructed from world knowledge the place targets are chosen from empirical measurements inside the organic matrix of curiosity. For this work, we wished to make use of strategies that may very well be totally acquired on the Q-LIT but nonetheless be able to detecting low-abundant peptides. One benefit of this strategy is that targets are tuned for the instrument from the context of retention time scheduling and optimum transition choice.
A typical limitation to workflows transitioning assays from high-resolution to nominal-mass accuracy devices (e.g., Q-LIT and triple quadrupoles) is the necessity to rebuild any given assay for the platform by re-validating transitions and peptides of curiosity. We applied a workflow and software program device to reap the benefits of the flexibility to generate peptide libraries utilizing off-line fractionated DDA or gas-phase fractionation data-independent acquisition (GPF-DIA), and developed software program to construct on-the-fly PRM assays for a similar instrument. (Fig. 2a). Briefly, the strategy used a DIA search engine to establish library peptides from GPF-DIA knowledge collected on the focused acquisition instrument with related acquisition parameters (particularly, 2 m/z isolation home windows) and the identical HPLC gradient used for PRM evaluation. The software program was applied to work with enter libraries from a beforehand generated high-resolution DDA-based spectrum library, peptide predictions utilizing Prosit, or direct DIA search engine outcomes (e.g., Chimerys). This evaluation translated current or predicted spectrum libraries for the PRM instrument by discovering when a given peptide elutes within the PRM gradient, figuring out instrument-specific fragmentation patterns, and figuring out which transitions stay quantitative when making low-resolution measurements. Moreover, the software program was designed to make use of retention instances from a just lately acquired DIA injection to extrapolate a schedule reflecting the present LC column situations. This assortment of PRM-validated peptides, known as a “translation library,” serves as a database of potential peptides to pick out for focused assays. Translation libraries act as DIA chromatogram libraries23,37 with the aim of effectively and rapidly translating the chemical traits of library entries for goal peptides from one instrument/acquisition strategy or prediction area to the measurement area of the instrument used for PRMs.

a A schematic for utilizing GPF-DIA to construct focused PRM assays. Quite a lot of library sources are first mined for library entries, then both re-searched utilizing EncyclopeDIA or inferred utilizing direct DIA serps, setting up a “translation” library of potential PRM targets and their chemical traits. Peptides marked for exclusion are faraway from the library, whereas the remaining peptides are sorted in keeping with their experimental relevancy (utilizing the third highest fragment). For this work, we had the software program construct PRM assays designed for 1, 10, and 100 ng enter ranges utilizing 10, 20, and 50 peptides per cycle, respectively. b Libraries had been generated utilizing both the interpretation library strategy or a extra commonplace DDA methodology coupled with offline high-pH reverse section (HPRP) fractionation. c The overlap of potential PRM candidates within the spectral library utilizing HPRP-DDA and the interpretation library filtered to a 1% peptide-level FDR.
The software program, mixed with the interpretation library, is designed to schedule a PRM assay from an inventory of goal accession numbers and different optionally desired accessions from a particular FASTA database. The assay might be modified utilizing each a peptide inclusion and exclusion record. Assays might be adjusted relying on instrument settings, the place the utmost assay density and a retention time scheduling window width have to be specified. Peptides are ordered based mostly on the third largest fragment ion per peptide, following widespread SRM/PRM conventions requiring at the very least three transitions11. The algorithm chooses peptides utilizing a grasping strategy, the place probably the most plentiful peptides are scheduled first based mostly on the right track preferences indicated by goal inclusion and exclusion lists. After the algorithm chooses a specified variety of peptides for a given protein (sometimes 3-5), no further peptides from that protein are thought of. Moreover, peptides can’t be added to a retention time area if any time level in that area has already reached the utmost assay density. As soon as the algorithm iterates by all potential peptides, the software program device produces a scheduling report and a goal inclusion record for the Thermo methodology editor. Whereas the device focuses on simplifying scheduling for Thermo devices, it’s analyzer and vendor-agnostic, supporting scheduling for each Orbitrap and ToF devices. This software program workflow has an accessible graphical consumer interface constructed into the EncyclopeDIA code base (see Supplementary Notice 2 and Supplementary Information 1 for extra particulars). Your complete workflow might be carried out in a single workday, from translation library to PRM assay. For this work, a single library was generated in 7.5 h, together with 6× h-long gradients adopted by 15 min for pattern loading and column equilibration, the place the interpretation library was processed in parallel with the acquisition.
To generate a low-input PRM assay on the Q-LIT, we examined two commonplace strategies of constructing libraries: a translation library utilizing GPF-DIA and a spectral library utilizing fractionated DDA. We collected libraries from a pool of IL-2 and IL-15-stimulated T cell proteomes. To construct the interpretation library, 6x gas-phase fractions had been used with 2 m/z broad isolation home windows throughout mass ranges of 100 m/z per injection. Because the background proteome matrix will not be drastically chemically altered or diluted, this strategy produces retention instances that carefully match the quantitative PRM experiments. In distinction, we offline fractionated the DDA library samples utilizing high-pH reverse section separations to yield a complete of 10 fractions, which had been analyzed in separate injections (Fig. 2b). Consequently, every fraction has a simplified matrix background, which can not mirror retention instances as constantly in unfractionated quantitative samples. The DDA and DIA strategies produced equally sized libraries, however surprisingly distinct populations of peptides (Fig. 2c), presumably because of the distinction in matrix complexity that resulted from the 2 fractionation strategies used to generate every library.
Along with producing barely extra peptide detections, peptide-centric extraction38 of DIA datasets is extra akin to fragment-level quantification utilizing focused strategies than DDA measurements39. As such, peptides detected utilizing GPF-DIA usually tend to produce strong, focused assays because the mode of discovery makes use of related methodologies to the ultimate quantitative measurements. Nonetheless, some pattern varieties, resembling enriched phosphopeptides, could also be higher suited to library era with DDA because the semi-stochastic sampling of precursor ions for fragmentation permits for a higher variety of distinctive positional isomers to be detected when combining technical replicates40. For this work, we proceeded with the interpretation library for assay growth, however the scheduling software program produced for this work capabilities with both a DDA or DIA-derived library supply.
For DIA injections, search outcomes from EncyclopeDIA and CHIMERYS had been mixed for downstream work. CHIMERYS is a spectrum-centric search engine that builds on INFERYS to supply spectra and retention-time predictions for peptides in a given FASTA database41. Compared, we searched a Prosit-predicted spectral library42,43 with the peptide-centric search engine EncyclopeDIA, tailored for analyzing ion lure knowledge. Consequently, EncyclopeDIA was restricted to looking out +2 and +3 peptides to take care of an affordable search area, whereas CHIMERYS was configured to think about modifications and better cost states. Extra peptides had been detected from CHIMERYS in comparison with EncyclopeDIA in every gas-phase fraction (Supplementary Fig. 1a), but contemplating this superset of detections elevated the entire variety of potential targets (Supplementary Fig. 1b). Each serps produced an equal variety of viable peptide targets that may very well be utilized in downstream PRM experiments (Supplementary Fig. 1c). In all circumstances, the retention instances from CHIMERYS-detected peptides had been re-peak picked utilizing EncyclopeDIA to establish candidate goal transitions for PRM measurement in a mixed DIA library.
Assessing Q-LIT PRM quantitative accuracy at low enter
With low-input world proteomics, we preferentially measure solely probably the most plentiful proteins. We stress-tested the quantitative accuracy of the Q-LIT system utilizing PRMs by measuring biologically related proteins that are inclined to happen at a spread of abundances within the proteome. To perform this, we first functionally annotated candidate peptides within the mixed DIA library utilizing the PANTHER database44. We chosen goal proteins based mostly on GO-terms and Reactome pathways for T cell differentiation, immune biology, T cell activation, cytokines, and transcription elements, specializing in deciding on proteins related to the dynamics of reminiscence or effector T cells. Utilizing the PRM scheduling algorithm, we constructed three assays utilizing the identical financial institution of proteins, the place every assay was suited to a special enter stage: as much as 50 peptides/cycle for 100 ng of fabric, 20 peptides/cycle for 10 ng of fabric, and 10 peptides/cycle for 1 ng of fabric. In the end, the 100 ng assay quantified 481 peptides, the ten ng assay quantified 151, and the 1 ng assay quantified 61. To take care of a 2-second cycle time utilizing 1 ng of fabric, the utmost ion injection time (maxIIT) was set to 200 ms. Equally, at 10 ng of fabric, the maxIIT was set to 95 ms (barely under 100 ms to accommodate the extra time required to route ions within the mass spectrometer). At 100 ng of fabric, the ion injection time was set to 50 ms; nonetheless, every scan not often met that size of time.
We carried out matrix-matched calibration curves45 at 100 ng, 10 ng, and 1 ng ranges to evaluate the quantitative accuracy of the Q-LIT over a number of orders of magnitude. Dilutions in a buffer background are useful to evaluate instrument sensitivity, however as a result of background noise decreases on the identical fee as goal peptides, quantitative linearity will at all times seem extra correct than in an actual background matrix. Matrix-matched calibration curves are more practical at assessing linearity in real-world situations because the background sign doesn’t change with dilution. To perform this, we needed to construct an appropriate background matrix of comparable composition to our goal T cell proteome. Our strategy used dimethyl labeling to change the foreground T cell proteome, protecting the identical composition whereas producing completely different precursor and fragment plenty. Dimethyl labeling was first launched as a multiplexing methodology the place a number of samples could be labeled and combined previous to mass spectrometry46. In our strategy, solely the background is modified, the place free amines are mass-shifted by two methyl teams (+28 Da). This shifts any labeled precursors (even incomplete reactions with a single methyl group) exterior the precursor isolation window utilized by PRM measurements, guaranteeing that foreground indicators won’t be confused with background indicators. Moreover, dimethyl labeling is inexpensive, straightforward, and fast, as peptides are labeled to 99.9% completion inside a 1-h response.
From this experiment, we discovered that cheap quantitative accuracy (Supplementary Fig. 2) might be achieved with the Q-LIT at low enter. At 100 ng, the quantitative accuracy of most peptides acquired with PRM stays constant for almost two orders of magnitude (Fig. 3a), the place the median decrease restrict of detection (LoD) was 0.83:100 (ratio of foreground to background) and the median decrease restrict of quantification (LoQ) was 2.8:100 (Fig. 3b), the place solely 0.6% of peptides couldn’t be assigned a LoQ. Quantification was barely worse on the 10 and 1 ng ranges, the place 4.6% and 20% of peptides couldn’t be assigned a LoQ (Fig. 3c). The anticipated goal ratios are annotated in Fig. 3d. Because the analyte sign dropped with reducing focus, background interference tended to overwhelm the analyte sign. In consequence, quantitative ratios with the Q-LIT are inclined to regress to 1:1, leading to higher-than-expected measured ratios. Unsurprisingly, on the 100 ng stage, the sign is extra simply distinguishable from noise, and the LoD distribution is usually decrease than at 10 ng or 1 ng (Fig. 3e and f).

a, b The quantitative accuracy of matrix-matched curves on an ion lure of pooled IL-2 and IL-15 peptides in a background of dimethyl-labeled peptides. We generated three curves loading 100 ng, 10 ng, and 1 ng of fabric on-column. Field plots present the unfold of measured values the place the whiskers point out 5% and 95% factors, and the daring line signifies the median measurement. d Every dilution is a special shade the place coloured dashed strains point out the anticipated fold change. e, f The distribution of the Figures of Benefit for the 1, 10, and 100 ng injections utilizing PRM on the Q-LIT. All boxplots (a–c, e, f) are represented as median worth. The field maxima extents to the first interquartile vary (twenty fifth percentile), whereas the minima extends to the third inside quartile vary (seventy fifth percentile).
We acquired analogous PRM assays on a Q-Orbitrap Exploris 480 at 1, 10, and 100 ng utilizing nearly all of peptides that had been quantified within the calibration curve assays for the Q-LIT. Q-Orbitraps have restricted trapping capability, which might restrict the dynamic vary inside a spectrum. In distinction to the Q-LIT, peptides with low background sign merely cease being measured earlier than they fall under the LoQ, leading to quantitative ratios regressing to 0:1 and decrease than anticipated measured ratios (Supplementary Fig. 3). A consequence of this sign drop-off is that estimated LoQ and LoD values for the Q-Orbitrap might be harder to estimate accurately, producing modes round samples the place the peptide sign falls under the spectrum dynamic vary (Supplementary Fig. 4). Though the distribution of LoDs stays related between the Q-LIT and Q-Orbitrap, the distribution of LoQs is usually decrease for the Q-LIT on a peptide-by-peptide foundation (Supplementary Fig. 5). Along with producing improved LoQs, extra peptides may very well be focused with the Q-LIT because the instrument has quicker scanning speeds. For instance, at 100 ng stage, 300 peptides had been scheduled on the Q-Orbitrap, whereas 473 peptides may very well be scheduled on the Q-LIT earlier than the schedule was at most capability.
Single cells sometimes produce between 0.1 and 0.3 ng of peptides, relying on the cell kind. Contemplating the 1 ng pattern, the median measured peptide produced a linear sign on this vary (0.198:1). A number of peptides confirmed a linear response under 0.1 ng. For instance, the peptide ECESYFK from Granzyme B was discovered to have a LoQ of 0.043:1, equating to a proteome fraction consisting of 43 pg in a background of 1 ng, and was nonetheless measurable above background on the 18 pg stage (Fig. 4a and b). Two different Granzyme B peptides, VAAGIVSYGYK and TQQVIPMVK, had produced even decrease LoDs (under 10 pg equivalents). Granzyme B is a serine protease implicated in a number of autoimmune illnesses47. All advised, 61 peptides with estimable LoQs within the 1 ng assay corresponded to 30 quantified proteins with a median of 11-14 factors throughout the height base utilizing the Q-Orbitrap and Q-LIT (Supplementary Fig. 6).

a Every row shows a peptide chromatogram at every dilution inside the 1 ng curve. Every peptide incorporates three consultant transitions. The primary peptide from Granzyme B had the perfect estimated LoQ at 0.043:1, whereas the third peptide from IL-2 receptor subunit alpha had an estimated LoQ at 0.132:1 at 1 ng. b LoQ and LoD had been estimated on a peptide-by-peptide foundation utilizing EncyclopeDIA’s curve becoming algorithm. First, the algorithm determines a most line by the noise of the calibration curve after which suits the linear dynamic vary. The intersection of each strains is the LoD (proven with a grey shaded, empty circle), whereas the LoQ (proven on the intersection of the dotted purple strains) is three commonplace deviations of the noise above the LoD. The error related to the strains fitted by the noise and linear, dynamic vary are proven in yellow, and symbolize 3 commonplace deviations above and under the median sign for every line.
Validated cell populations for quantitative testing
Along with exhibiting quantitative accuracy in a managed matched matrix, we wished to validate measurement precision in low-input organic experiments. The interleukins (IL) household of proteins is a category of cytokines expressed by many cells, together with immune cells, which bind to particular receptors that elicit pro- and anti inflammatory roles48. Sure cytokines, resembling IL-2 and IL-15, bind to receptors on the floor of T cells in particular organic occasions, resembling activation and differentiation. Each of those molecules have been efficiently used as a part of immunotherapies to fight most cancers49,50,51,52. Apparently, IL-2 and IL-15 are structurally related in homology and activate T cells by the identical receptor subunits (IL-2/IL-15Rβγ)53,54, mediating largely related organic results on T cells55,56. Nonetheless, probably associated to the expression of the non-public IL-2Rα and IL-15Rα chains, IL-2 induces an effector-like phenotype (with low CD62L expression) whereas IL-15 induces a memory-like phenotype (with greater CD62L)57. We generated activated T cells cultured in IL-2 or IL-15 to duplicate an effector-like and memory-like phenotype for CD4+ and CD8+ cells (Supplementary Fig. 7a). We chosen this mannequin system to showcase the flexibility to generate LIT-PRM assays utilizing well-studied biology at inputs under 1 ng. Moreover, movement cytometry was used as an orthogonal method to validate the cell populations current in IL-2 and IL-15 handled T cells on days 5, 6, and 10, which exhibited an effector-like and memory-like phenotype (Supplementary Fig. 7b and c).
At day 10, movement cytometry recognized that every tradition was predominantly composed of T cells, with CD8+ T cells being the bulk subset in each IL-2 (83.8%) and IL-15 (92.2%) cultures (Fig. 5). Correspondingly, we discovered that CD4+ T cells composed 14.6% of the cells stimulated with IL-2 and 6.9% of the cells stimulated by IL-15. This was mirrored in our focused proteomics knowledge, because the CD4 protein was the second most downregulated protein in IL-15-stimulated T cells in comparison with IL-2-stimulated cells (Fig. 6a). We notice that this protein was not technically quantified at 1 ng, because the one peptide for CD4 (VVQVVAPETGLWQCLLSEGDKVK) lacked linearity in sign as estimated by the calibration curve. We measured the identical peptide on the 10 ng stage, the place we calculated the LoD to be 0.96:10 (ratio of foreground to background) with an LoQ of 8.3:10 (Supplementary Fig. 8). This indicated that on the 1 ng stage, CD4 needs to be above the LoD however under the LoQ, and our outcomes match these calculations.

a The gating process was used to find out the relative proportion of every cell kind within the IL-2 and IL-15 samples (extra particulars in Supplementary Fig. 7). b The estimated cell populations based mostly on again calculations of the gating outcomes.

a Quantitative ratios for the panel proteins assayed within the 10 peptide/cycle PRM. The assay was collected in technical triplicate injections of Day 10 IL-2 and IL-15-stimulated T cell proteomes. The chosen panel of proteins is related to T cell activation, differentiation, or cytokine signaling. No LoQ was decided for CD4 with the 1 ng calibration curve, indicated by a purple 5-point star. Within the IL-2 stimulated pattern, IL2RB was measured under the LoQ decided by the 1 ng calibration curve, indicated by a 6-point star. The 9 knowledge factors of every protein had been extracted from 3 technical replicate PRM injections for the IL-2 and IL-15 stimulated proteomes by calculating the log2 fold change in all potential combos utilizing technical replicates. The boxplot facilities are represented as median values. For every field, the maxima extents to the first interquartile vary (twenty fifth percentile) and the minima extends to the third inside quartile vary (seventy fifth percentile. b Coefficient of technical variation (% CV) plots for all peptides quantified within the 1 ng assay. The purple dotted line signifies 20% CV on every plot.
IL-2 stimulation is thought to push activated T cells into an effector-like inhabitants, which is mirrored by the paired movement cytometry knowledge on day 10 of remedy with recombinant human IL-2. Granzyme B, from which we estimated probably the most responsive peptide (TQQVIPMVK) was quantitative to 0.025:1 (ratio of foreground to background), is an effector molecule secreted by cytotoxic CD8+ T cells. We discovered peptides related to this protein had been 1.55x decrease in IL-15 than IL-2 stimulated cells utilizing focused proteomics (Fig. 6a), which matches movement cytometry knowledge indicating that the variety of effector CD8+ T cells (TEFF) is decrease when stimulated with IL-15 than IL-2. Whereas each IL-2 and IL-15 resulted in T cell activation, IL-15 stimulation differentiated memory-like T cells, as demonstrated within the movement cytometry knowledge. The CD44 receptor antigen is a cell floor receptor that helps cells facilitate cell-cell interplay and response to the tissue microenvironment. Apparently, we discovered that the expression of CD44 is barely greater in IL-2 in comparison with IL-15, indicating that IL-2 stimulated cells had the next inhabitants of activated cells, with a 1.6x median fold change in abundance. T cells that specific CD62L have an elevated inhabitants of reminiscence T cells after IL-15 stimulation54. Movement cytometry knowledge indicated that we had the next inhabitants of CD62L+ cells within the IL-15 stimulated situation than T cells stimulated with IL-2 (Supplementary Fig. 7), specifying the next inhabitants of memory-like T cells. In comparison with the IL-15 stimulated cells, IL-2 stimulated T cells expressed IL-2Rβ/IL-15Rβ at the next ratio (Fig. 6a), which is related to a reminiscence phenotype. Generally, we observe excessive analytical precision utilizing PRM with a Q-LIT platform, even in 1 ng assays. The vast majority of peptides had been measured with lower than a 20% coefficient of variation between 3 technical replicates (Fig. 6b), indicating quantitative rigor inside the workflow.
In the end, we detected 100% of the proteins monitored with movement cytometry utilizing world proteomics throughout library era. Whereas a few of these proteins had been exhausting to look at at low enter (1 ng), we had been in a position to quantify 75% above an estimated LoQ with focused proteomics. This overlap signifies complementary advantages of utilizing movement cytometry in tandem with focused proteomics to seize the immune cell state totally. Whereas single-cell proteomics utilizing mass spectrometry continues to develop, movement cytometry is the perfect methodology for measuring a small variety of proteins (6-12) on 1000’s of particular person cells inside a single day. Alternatively, focused mass spectrometry on 1-10 T cells (equal to round 0.1 and 1 ng) can monitor tens to tons of of proteins, together with cytokines and transcription elements, which can’t be simply monitored utilizing movement cytometry.